Прогерия или известна още като синдром на Хътчинсън-Гилфорд е 1 от 10-тте най-редки заболявания.
Чудатите тела, за които не знаем много.
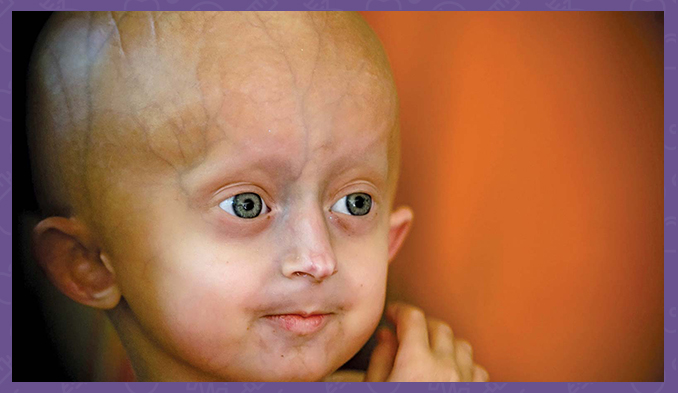
Прогерия е едно от най-редките генетични заболявания. В света има регистрирани около 100 случая на болестта. Характеризира се с изключително бързото състаряване на тялото.
Синдромът на Хатчинсън-Гилфорд е генетично състояние, характеризиращо се с драматичната, бърза поява на стареене в началото на детството. Засегнатите деца обикновено изглеждат нормални при раждането и в ранна детска възраст, но след това растат по-бавно от другите деца и не наддават на тегло с очакваното темпо. Те развиват характерен външен вид на лицето, включително изпъкнали очи, тънък нос със заострен връх, тънки устни, малка брадичка и стърчащи уши. Прогерия или синдромът на Хътчинсън-Гилфорд също причинява загуба на коса (алопеция), застаряване на кожата, аномалии в ставите и загуба на мазнини под кожата (подкожна мастна тъкан). Това състояние не засяга интелектуалното развитие или развитието на двигателни умения като седене, стоене и ходене.
Характеристика
Хората с Прогерията изпитват силно втвърдяване на артериите (артериосклероза) в началото на детството. Това състояние значително увеличава шансовете за сърдечен удар или инсулт в млада възраст. Тези сериозни усложнения могат да се влошат с времето и са животозастрашаващи за засегнатите хора. Състояние е много рядко. Съобщава се, че се среща при 1 на 4 милиона новородени по целия свят.
Синдромът на Хатчинсън-Гилфорд / прогерията се счита за автозомно доминантно състояние, което означава, че едно копие на променения ген във всяка клетка е достатъчно, за да причини разстройството. Състоянието е резултат от нови мутации в гена на LMNA и почти винаги се среща при хора без анамнеза за подобно нарушение в семейството им.
Прогерията обикновено не се предава в семействата. Промяната на гена почти винаги е случайно явление, което е изключително рядко. Децата с други видове прогероидни синдроми, които не са HGPS/ Синдромът на Хатчинсън-Гилфорд/, могат да имат заболявания, които се предават в семействата. HGPS обаче се дължи на спорадична автозомно доминантна мутация. Спорадична, защото това е нова промяна в това семейство и доминираща, защото само едно копие на гена трябва да бъде променено, за да има синдром. За родителите, които никога не са имали дете с прогерия, шансовете да имат дете с прогерия са 1 на 4 – 8 милиона, но за тези, които вече са имали дете с прогерия, шансът се увеличава до около 2-3%. Това се дължи на състояние, наречено мозаицизъм, при което родител има генетична мутация за прогерия в малка част от своите клетки, но няма прогерия.
Диагностициране
Прогерия обикновено се диагностицира през втората година от живота на детето или по-късно, когато прогероидните характеристики започват да се забелязват. Диагнозата се основава на задълбочена клинична оценка, характерни физически находки, внимателна анамнеза на пациента и диагностично генетично изследване. По-рядко може да се подозира наличието на синдрома още при раждане, въз основа на разпознаване на някои съмнителни проявления.
За правилната анамнеза могат да се провеждат специализирани образни тестове за потвърждаване или характеризиране на някои скелетни аномалии, потенциално свързани с болестта, като:
- дегенеративни промени (остеолиза) на някои кости на пръстите (терминални фаланги)
- и/или тазобедрената муфа (ацетабулум).
Диагнозата допълват задълбочени сърдечни оценки и текущ мониторинг (например клинични прегледи, рентгенови изследвания, специализирани сърдечни тестове) за оценка на свързаните сърдечно-съдови аномалии и определяне на подходящо управление на заболяването.
Видове терапии. Лечение
През септември 2012 г. резултатите от първото по рода си клинично изпитване за лекарства за деца с прогерия разкриха, че Lonafarnib, вид инхибитор на фарнезилтрансфераза (FTI), първоначално разработен за лечение на рак, е показан като ефективен за прогерия. Децата, приемащи препарата, показват подобрение в един или повече от един измежду четирите основни параметъра:
- покачване на допълнително тегло
- по-добър слух
- подобрена структура на костите
- и най-важното – повишена гъвкавост на кръвоносните съдове
Освен lonafarnib, който не е одобрен от FDA и по този начин се предлага само чрез клинични изпитвания на лекарства, лечението на HGPS (Hutchinson–Gilford progeria syndrome) е насочено към специфичните симптоми, които са очевидни при всеки засегнат индивид. Управлението на заболяването може да изисква координираните усилия на екип от специалисти, на които може да се наложи систематично и цялостно да планират лечението на засегнатото дете. Екипът може да включва:
- педиатри
- лекари, които диагностицират и лекуват нарушения на скелета, мускулите, ставите и други свързани тъкани (ортопеди)
- лекари, които диагностицират и лекуват аномалии на сърцето и основните му кръвоносни съдове
- физически терапевти
За повече информация, ние, Медикъл Караджъ сме на ваше разположение.
Обадете ни се на телефоните на „Медикъл Караджъ“: 0879 977 401 или 0879 977 402.
Следете и постоянно обновяващото ни се съдържание във Фейсбук.








