Progeria or also known as Hutchinson-Gilford syndrome is 1 of the 10 rarest diseases.
The weird bodies we don't know much about.
Progeria is one of the rarest genetic diseases. There are about 100 registered cases of the disease worldwide. It is characterized by extremely rapid aging of the body.
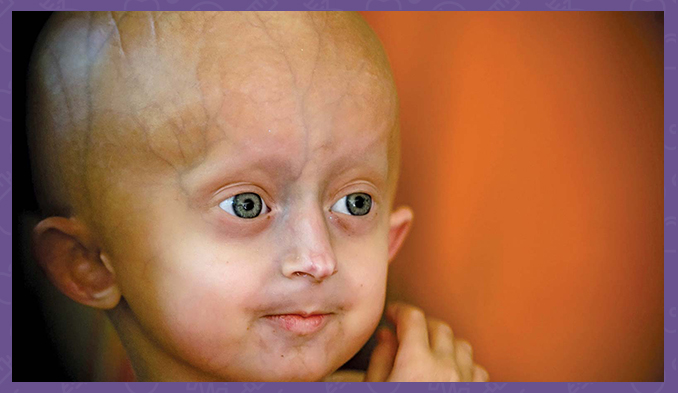
Hutchinson-Gilford syndrome is a genetic condition characterized by the dramatic, rapid onset of aging in early childhood. Affected children usually appear normal at birth and in infancy, but then grow more slowly than other children and do not gain weight at the expected rate. They develop a characteristic facial appearance, including bulging eyes, a thin nose with a pointed tip, thin lips, a small chin and protruding ears. Progeria, or Hutchinson-Gilford syndrome, also causes hair loss (alopecia), aging skin, joint abnormalities, and loss of fat under the skin (subcutaneous fat). This condition does not affect intellectual development or the development of motor skills such as sitting, standing and walking.
Feature
People with Progeria experience severe hardening of the arteries (arteriosclerosis) in early childhood. This condition greatly increases the chances of having a heart attack or stroke at a young age. These serious complications can worsen over time and are life-threatening for affected people. The condition is very rare. It is reported to occur in 1 in 4 million newborns worldwide.
Hutchinson-Gilford syndrome/progeria is considered an autosomal dominant condition, meaning that one copy of the altered gene in each cell is enough to cause the disorder. The condition results from new mutations in the LMNA gene and almost always occurs in people with no family history of such a disorder.
Progeria is not usually transmitted in families. Gene alteration is almost always a random event, which is extremely rare. Children with other types of progeroid syndromes that are not HGPS/ Hutchinson-Gilford syndrome/ can have diseases that are passed in families. HGPS, however, is due to a sporadic autosomal dominant mutation. Sporadic because it is a new change in that family and dominant because only one copy of the gene needs to be changed to have the syndrome. For parents who have never had a child with progeria, the odds of having a child with progeria are 1 in 4 - 8 million, but for those who have already had a child with progeria, the odds increase to about 2-3%. This is due to a condition called mosaicism, in which a parent has the genetic mutation for progeria in a small proportion of their cells but no progeria.
Diagnosing
Progeria is usually diagnosed in the second year of a child's life or later, when progeroid features begin to become noticeable. Diagnosis is based on a thorough clinical evaluation, characteristic physical findings, a careful patient history, and diagnostic genetic testing. Less commonly, the presence of the syndrome may be suspected at birth, based on recognition of certain suspicious manifestations.
For a proper history, specialized imaging tests can be performed to confirm or characterize certain skeletal abnormalities potentially associated with the disease, such as:
- degenerative changes (osteolysis) of some finger bones (terminal phalanges)
- and/or hip socket (acetabulum).
Diagnosis is complemented by thorough cardiac evaluations and ongoing monitoring (e.g., clinical examinations, x-rays, specialized cardiac tests) to assess associated cardiovascular abnormalities and determine appropriate disease management.
Types of therapy. Treatment
In September 2012, the results of the first-ever clinical trial for a childhood progeria drug revealed that Lonafarnib, a type of farnesyltransferase inhibitor (FTI) originally developed to treat cancer, was shown to be effective for progeria. Children taking the drug showed improvement in one or more of one among four key parameters:
- extra weight gain
- better hearing
- improved bone structure
- and most importantly - increased flexibility of blood vessels
Aside from lonafarnib, which is not FDA approved and thus only available through clinical drug trials, the treatment of HGPS (Hutchinson-Gilford progeria syndrome) targets the specific symptoms that are evident in each affected individual. Management of the disease may require the coordinated efforts of a team of professionals who may need to systematically and comprehensively plan the treatment of the affected child. The team may include:
- pediatricians
- doctors who diagnose and treat disorders of the skeleton, muscles, joints and other related tissues (orthopaedists)
- doctors who diagnose and treat abnormalities of the heart and its major blood vessels
- physical therapists
For more information, we at Medical Karaj are at your service.
Call us on the following numbers "Medical Karaj": 0879 977 401 or 0879 977 402.
Also keep an eye on our constantly updated Facebook content.








